La Polidattilia Ulnare è l’anomalia congenita delle mani più riscontrata nei bambini afroamericani. Si verifica in circa 1 su 150 neonati. Sebbene questa condizione sia molto meno comune nei caucasici (1: 1.400), messicani (1: 700) e mediorientali (1:1.000), è tuttavia una delle anomalie congenite più frequentemente riscontrate dell’arto superiore. Esistono differenti associazioni di sottotipi di polidattilia ulnare in bambini di diversa razza.
La polidattilia ulnare sec. Tetamy e McKusick va distinta in tipo A e B. La polidattilia ulnare tipo A presenta un dito accessorio completamente sviluppato che si articola con il quinto metatarso (piede) o metacarpo (mano) o con un quinto metatarso (piede) o metacarpo (mano) duplicato. include dita più sviluppate con anatomia variabile. Contengono sempre elementi ossei e possono presentare tendini flessori ed estensori anomali. Le articolazioni interfalangee sono spesso ipoplasiche e rigide. Le dita di tipo A contengono sempre nervi, arterie e vene digitali.
ll dito accessorio di tipo B è di tipo rudimentale-vestigiale e privo di componenti ossee e si manifesta frequentemente solo come escrescenza cutanea. Tuttavia, questo schema non distingue la gamma di forme intermedie che richiede un’ulteriore classificazione in cinque specifici pattern morfologici, basati sul grado di duplicazione del metatarso (parte centrale del raggio di un piede). Dal meno differenziato al più differenziato i tipi sono i seguenti:
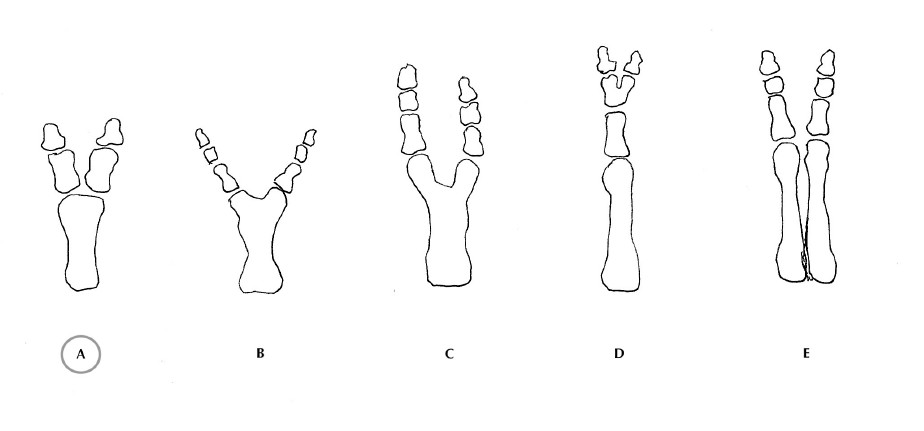
B: V metatarso a forma di T
C: metatarso a forma di Y
D: polidattilia parziale
E: duplicazione completa
La forma più comune è il tipo A, duplicazione della falange prossimale con una testa metatarsale larga.
Nei bambini afroamericani, la condizione è generalmente ereditata in maniera autosomica dominante, ed è raramente associata ad altre anomalie della mano, sindromi congenite o anomalie sistemiche non sindromiche. La polidattilia ulnare dei bambini afroamericani è solitamente di tipo B e generalmente bilaterale (70%). Nei casi unilaterali la mano sinistra risulta più colpita. C’è una distribuzione omogenea tra sessi.
Nei pazienti di origine non africana, la polidattilia ulnare è solitamente sporadica. Solo il 5% dei pazienti dimostra un modello riconoscibile di ereditarietà. I pazienti non africani sono affetti sia da polidattilia ulnare di tipo A che da quella di tipo B. Nel 20% dei casi è bilaterale e i maschi sono più comunemente colpiti delle donne. I pazienti non africani dimostrano anche
una maggiore incidenza di condizioni associate alle mani tra cui polisindattilia, polidattilia mista e sindattilia isolata rispetto ai bambini afroamericani. In alcuni bambini può anche essere presente coinvolgimento del piede (cioè polidattilia bilaterale dei piedi).
Prima



Dopo


Prima



Dopo


Prima

Dopo



I pazienti con polidattilia ulnare possono presentare associazioni con altre
sindromi congenite come:
- Ellis-van Crevald syndrome
- Smith-Lemli-Opitz
- McKusick-Kaufman syndrome
- Trisomy 13 Patau syndrome
- Short rib-polydactily I-III
- Orofaciodigital syndrome
- Bardet-Biedel syndrome
- Meckel syndrome
- Greig cephalopolysyndactily syndrome
- Pallister Hall syndrome
- Joubert syndrome
- Simpson-Galebi-Behmel syndrome
- Hydrolethalus syndrome
- Acrocallosal syndrome
- Asphyxiating thoracic dystrophy/Jeune syndrome
- Focal dermal hypoplasia/Goltz-Gorlin syndrome
Anche la polidattilia ulnare isolata non-sindromica è fortemente associata ai modelli di ereditarietà genetica. Sono stati descritti entrambi i pattern autosomici dominanti e recessivi. L’analisi genetica ha collegato la polidattilia ulnare ai cromosomi 7, 13 e 19. Tuttavia, il modello esatto di ereditarietà è incerto e molto probabilmente più complesso rispetto ad una trasmissione mendeliana. Esiste anche un rischio legato all’esposizione ambientale per lo sviluppo della malformazione. Inoltre è stata collegata al fattore di trascrizione Gli-3, una proteina importante localizzata sul cromosoma 7, che esiste in una forma attiva (Gli-3A) e una forma inattiva (Gli-3R).
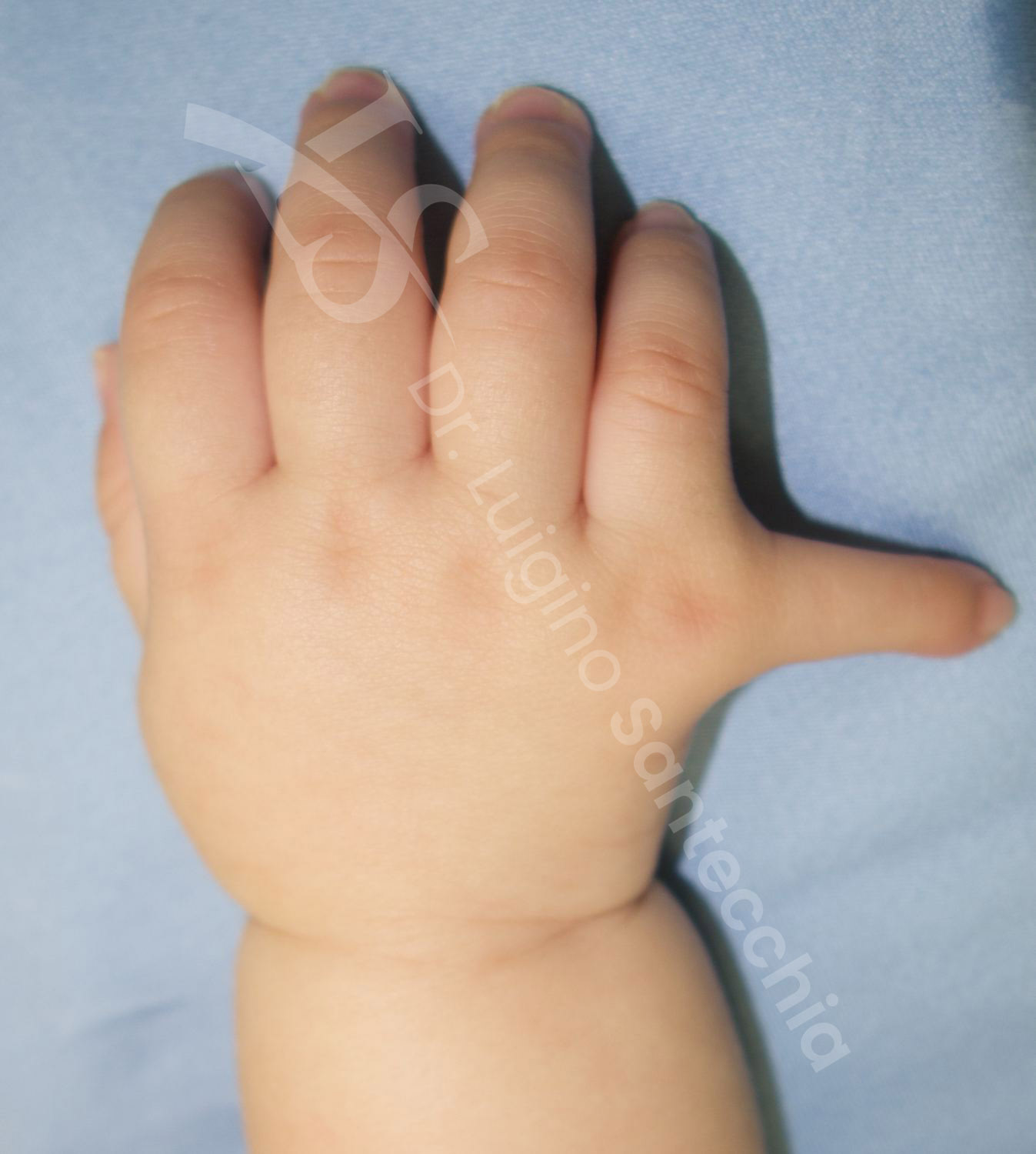
Clinicamente La polidattilia ulnare tipo B include dita rudimentali sovrannumerarie che originano dal bordo ulnare del mignolo. Poiché queste dita mancano di elementi ossei e tendini, non sono funzionali. I remnant possono essere piccoli come una protuberanza simile a una verruca sul lato ulnare della falange prossimale del mignolo, oppure possono assumere la forma di una piccolo dito in miniatura con un ossicino fibro-cartilagineo e un’unghia ipoplasica (dito pendulo). Le piccole protuberanze della polidattilia rudimentale vanno interpretate come monconi residui di dita che hanno subìto amputazione in utero. Tutte le dita sovrannumerarie di tipo B, compresa la polidattilia rudimentale, contengono un peduncolo neurovascolare.
Diagnosi
La polidattilia ulnare, in particolare il tipo A, può essere diagnosticata in corso di ecografia prenatale di routine al secondo trimestre. I restanti casi dovrebbero essere diagnosticati durante gli esami fisici postnatali di routine. La polidattilia rudimentale viene diagnosticata mediante un attento esame della cute sul bordo ulnare della mano. L’esame obiettivo dovrebbe concentrarsi su elementi associati a sindromi incluse nell’elenco precedente. Nei bambini con polidattilia ulnare di tipo A, le radiografie antero-posteriori aiuteranno a chiarire
l’anatomia ossea dell’elemento soprannumerario. La storia familiare deve essere raccolta oculatamente per determinare una potenziale causa genetica. Il riferimento a un genetista deve essere considerato nei pazienti con evidenza di sindromi congenite o polidattilia familiare ulnare isolata.
I genitori dei pazienti con polidattilia ulnare di tipo A rimangono spesso sorpresi dalla differenza anatomica della mano del loro bambino. Si deve considerare che il trattamento non rappresenta un’emergenza. Molti neonati con polidattilia ulnare di tipo B sono trattati con legatura chirurgica in reparto neonatale da parte del personale infermieristico, pediatri, ostetrici e intensivisti neonatali. In breve tempo l’appendice digitale diventa ischemica e cade dopo giorni o settimane dopo l’applicazione della legatura. Per lo stesso trattamento
possono essere impiegate anche le clips vascolari. Sono riportati casi di pazienti non trattati, in cui l’appendice può auto-amputarsi e cadere senza l’intervento.
In via del tutto teorica queste appendici pendule possono costituire un pericolo, dal momento che i neonati portano spesso la mano in bocca, con rischio che il peduncolo si stacchi e l’elemento possa venire inalato accidentalmente.
In attesa di trattamento, consigliamo sempre di coprire con cerotto molto aderente, onde scongiurare questo raro ma grave pericolo.
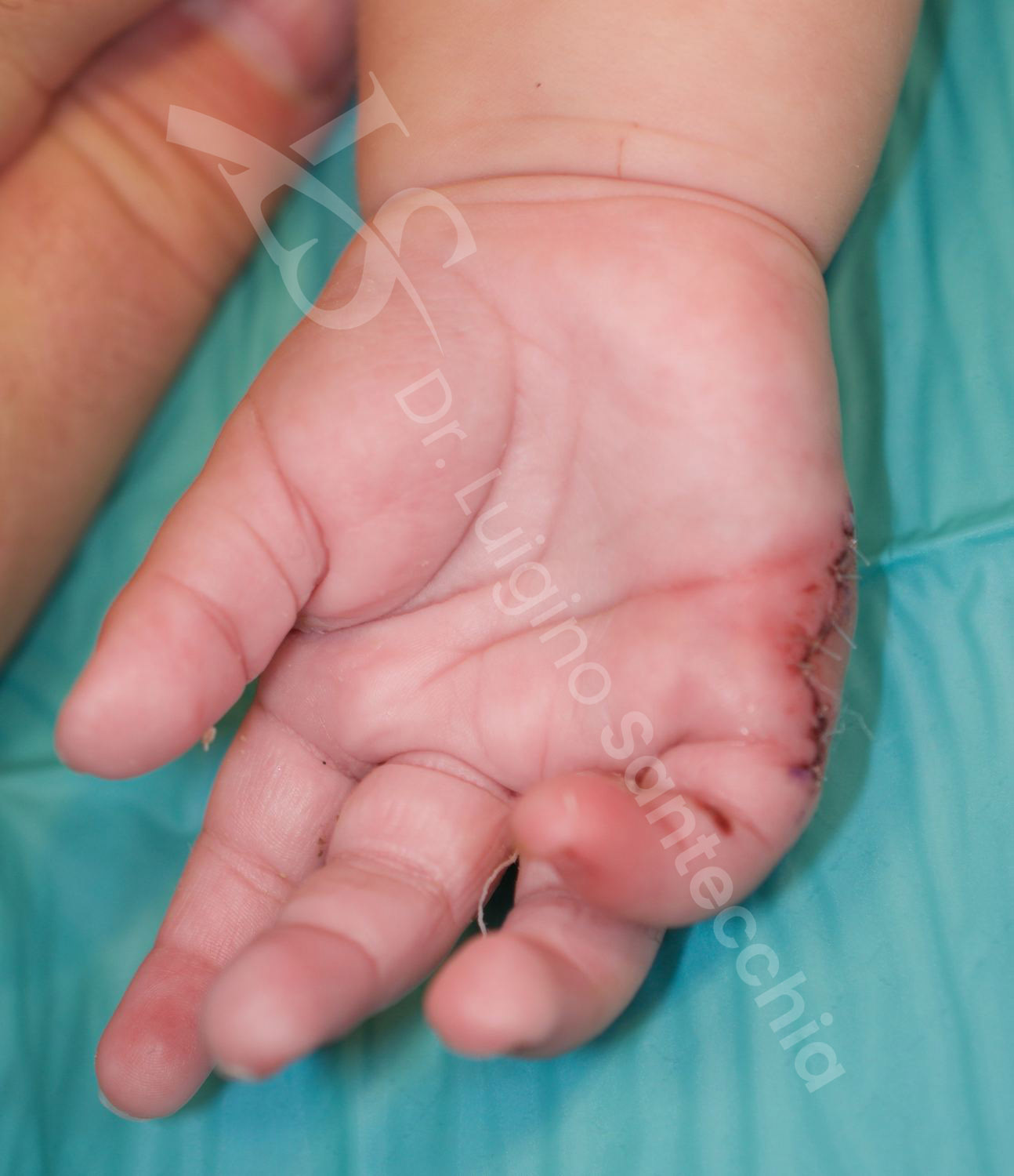
I neonati con polidattilia ulnare completamente sviluppata sono di solito riferiti a un chirurgo della mano, che provvede al trattamento di competenza, idealmente tra gli 8 e i 18 mesi di vita.
In via del tutto teorica queste appendici pendule possono costituire un pericolo, dal momento che i neonati portano spesso la mano in bocca, con rischio che il peduncolo si stacchi e l’elemento possa venire inalato accidentalmente.
In attesa di trattamento, consigliamo sempre di coprire con cerotto molto aderente, onde scongiurare questo raro ma grave pericolo.
I neonati con polidattilia ulnare completamente sviluppata sono di solito riferiti a un chirurgo della mano, che provvede al trattamento di competenza, idealmente tra gli 8 e i 18 mesi di vita.
Complicanze
Le complicanze correlate al trattamento sono sorprendentemente più frequenti nella legatura piuttosto che nell’escissione chirurgica. Infatti se la sutura non viene posta esattamente alla base del peduncolo, la cute residua prossimale può persistere come una protuberanza visibile e palpabile (40% dei casi); nel sito di amputazione può inoltre formarsi un neuroma doloroso. Questa complicanza viene ridotta al minimo mediante escissione chirurgica con neurectomia da trazione. Altre complicanze associate a una legatura malposizionata includono il sanguinamento, l’ematoma, l’infezione e la necrosi senza amputazione.
Le complicanze correlate al trattamento della polidattilia di tipo A sono rare. Comprendono il sanguinamento e l’ematoma postoperatorio, ma possono essere ridotte al minimo con un’operazione ben pianificata ed eseguita. La ricostruzione nella polidattilia ulnare tende a determinare instabilità o rigidità articolare, meno sintomatica rispetto alla polidattilia radiale. Tuttavia, sono segnalati casi con prominenza della testa metacarpale residua delle piccole dita, e instabilità dell’articolazione metacarpo-falangea.